
A study published in Nature Communications shows that SMA compromises the development of the central nervous system from the embryonic stage, highlighting the importance of early therapeutic intervention
Spinal Muscular Atrophy (SMA), a genetic disorder traditionally considered a neurodegenerative disease of motor neurons, actually begins much earlier, already in the very first stages of embryonic development. This is the key finding of a study recently published in Nature Communications, resulting from a collaboration between Humanitas Research Hospital, Humanitas University, the Dino Ferrari Center of the University of Milan, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, and Columbia University in New York.
The research was coordinated by Simona Lodato – Professor of Human Anatomy at Humanitas University and group leader of the Neurodevelopment Laboratory at Humanitas Research Hospital, and by Stefania Corti – Full Professor of Neurology at the University of Milan and Director of the Neuromuscular and Rare Diseases Unit at the Policlinico of Milan. The findings redefine the understanding of SMA, demonstrating that the disease does not only involve degeneration of mature motor neurons, but disrupts the developmental programs of the central nervous system from the very beginning.
Organoids to study the early stages of the disease
To reach these conclusions, researchers used cerebral and spinal organoids – three-dimensional models derived from patient cells that are able to recapitulate key stages of human neuronal development in vitro. This approach allowed to directly observe the effects of SMN protein deficiency – the underlying cause of SMA – during the early phases of neuronal differentiation.
“We discovered that reduced SMN profoundly alters differentiation programs already at the level of neural progenitors,” explains Stefania Corti. “The disease therefore begins long before the appearance of clinical symptoms. This strongly reinforces the importance of newborn screening and early initiation of therapy.”
Single-cell sequencing analyses performed on the organoids revealed widespread alterations affecting several neuronal populations, not only motor neurons. These changes are accompanied by functional abnormalities: electrophysiological recordings showed pathological hyper-excitability in both spinal and cerebral organoids, indicating that the dysfunction involves the entire central nervous system.
“Organoids allowed us to observe biological processes that were previously inaccessible,” emphasizes Simona Lodato. “We were able to follow in real time how the lack of SMN interferes with neuronal developmental trajectories, creating a block in cell maturation. These models are essential not only for understanding the disease, but also for evaluating new therapeutic strategies in a human context.”

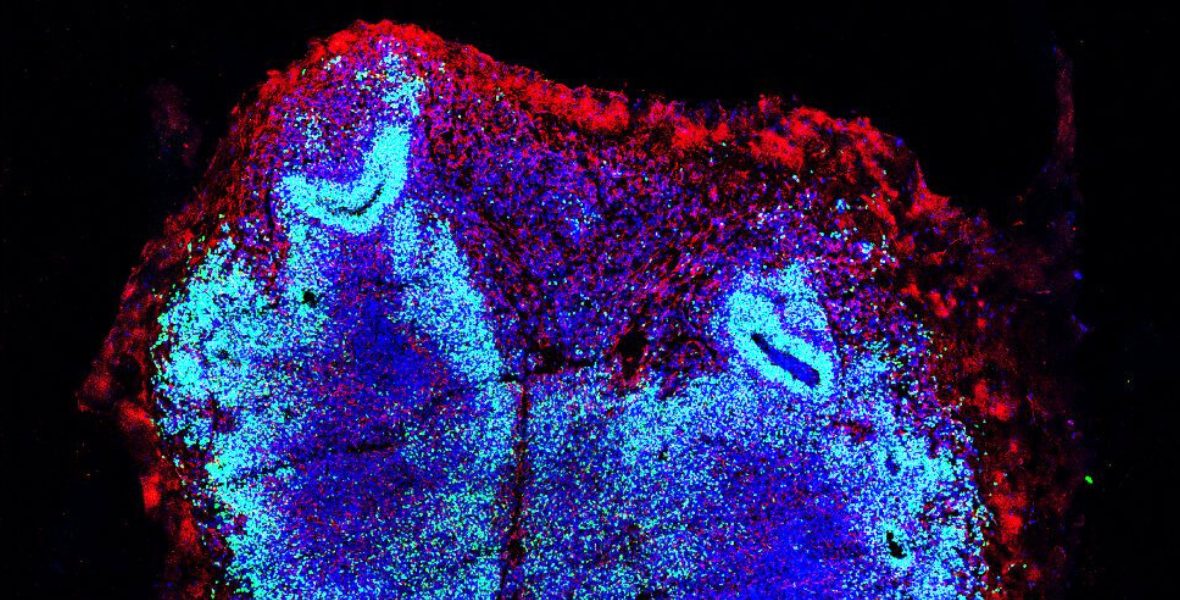
Spinal organoid sections stained with markers of neuronal progenitors (labeled in green) and neurons (red) from healthy control and SMA patients showing alterations in the SMA neurodevelopmental trajectories, indicating that this neurodegenerative disorder is caused by early defects in the spinal cord and central nervous system development. Nuclei were stained in blue. Scale bar: 200 or 100 μm.
Figure modified from Faravelli, I., Rinchetti, P., Tambalo, M. et al., Nature Communications 2026.
The value of early therapeutic intervention
Another key outcome of the study concerns the effectiveness of early treatment. The authors demonstrate that timely administration of antisense oligonucleotides, a therapy designed to increase SMN protein levels, can correct the morphological and functional defects observed in the early stages of development. The treatment restores physiological patterns of electrical activity and significantly reduces cell death, providing strong experimental evidence for the importance of intervention before neurological damage becomes irreversible.
Among the senior authors is also Monica Nizzardo, a researcher at the Dino Ferrari Center, who contributed her expertise in translational SMA research. The study is the result of a synergistic effort involving numerous researchers, including three young scientists as first authors: Monica Tambalo (a researcher in Professor Lodato’s group at Humanitas University and Humanitas Research Hospital), Irene Faravelli, and Paola Rinchetti, who made decisive contributions to the development and analysis of organoid models and to the interpretation of molecular and functional data. The publication represents the culmination of a multi-year collaboration among leading institutions in Italian neuroscience, integrating basic, clinical, and translational research.
Spinal Muscular Atrophy remains one of the leading genetic causes of infant mortality, but therapies introduced in recent years have already transformed the natural history of the disease. This study adds a fundamental piece to the understanding of its mechanisms, indicating that the greatest benefits are achieved when treatment is initiated in the presymptomatic phase, ideally through newborn screening, before alterations in nervous system development become irreversible.





