
Uno studio pubblicato su Nature Communications dimostra che la SMA compromette lo sviluppo del sistema nervoso centrale fin dalle fasi embrionali, evidenziando l’importanza di un intervento terapeutico precoce.
L’Atrofia Muscolare Spinale (SMA), patologia genetica tradizionalmente considerata una malattia neurodegenerativa dei motoneuroni, inizia in realtà molto prima, già nelle primissime fasi dello sviluppo embrionale. È quanto emerge da uno studio appena pubblicato su Nature Communications, frutto di una collaborazione tra l’IRCCS Istituto Clinico Humanitas, Humanitas University, il Centro Dino Ferrari dell’Università degli Studi di Milano, la Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico e la Columbia University di New York.
La ricerca è stata coordinata Simona Lodato – Professoressa di Anatomia Umana presso Humanitas University e group leader del laboratorio di Neurosviluppo dell’IRCCS Istituto Clinico Humanitas, e da Stefania Corti – Professoressa Ordinaria di Neurologia all’Università degli Studi di Milano e Direttrice della SSD Malattie Neuromuscolari e Rare del Policlinico di Milano. I risultati ridefiniscono la visione della SMA, dimostrando che la malattia non riguarda solo la degenerazione dei motoneuroni maturi, ma compromette fin dall’inizio i programmi di sviluppo del sistema nervoso centrale.
Organoidi per studiare le fasi precoci della malattia
Per arrivare a queste conclusioni, i ricercatori hanno utilizzato organoidi cerebrali e spinali: modelli tridimensionali derivati da cellule di pazienti, capaci di ricapitolare in vitro fasi chiave dello sviluppo neuronale umano. Questo approccio ha permesso di osservare direttamente gli effetti della carenza della proteina SMN – la cui mancanza è alla base della SMA – nelle fasi iniziali del differenziamento neuronale.
«Abbiamo scoperto che la riduzione di SMN altera profondamente i programmi di differenziamento già a livello dei progenitori neurali», spiega Stefania Corti. «La malattia, quindi, inizia molto prima della comparsa dei sintomi clinici. Questo rafforza in modo significativo l’importanza dello screening neonatale e dell’avvio precoce delle terapie».
Le analisi di sequenziamento a singola cellula condotte sugli organoidi hanno rivelato alterazioni diffuse che coinvolgono diverse popolazioni neuronali, non solo i motoneuroni. A queste si associano anomalie funzionali: le registrazioni elettrofisiologiche hanno infatti evidenziato una ipereccitabilità patologica sia negli organoidi spinali sia in quelli cerebrali, indicando che la disfunzione interessa l’intero sistema nervoso centrale.
«Gli organoidi ci hanno consentito di osservare processi biologici finora inaccessibili», sottolinea Simona Lodato. «Abbiamo potuto seguire in tempo reale come la mancanza di SMN interferisca con le traiettorie di sviluppo neuronale, creando un blocco nella maturazione delle cellule. Si tratta di modelli fondamentali non solo per comprendere la malattia, ma anche per valutare nuove strategie terapeutiche in un contesto umano».

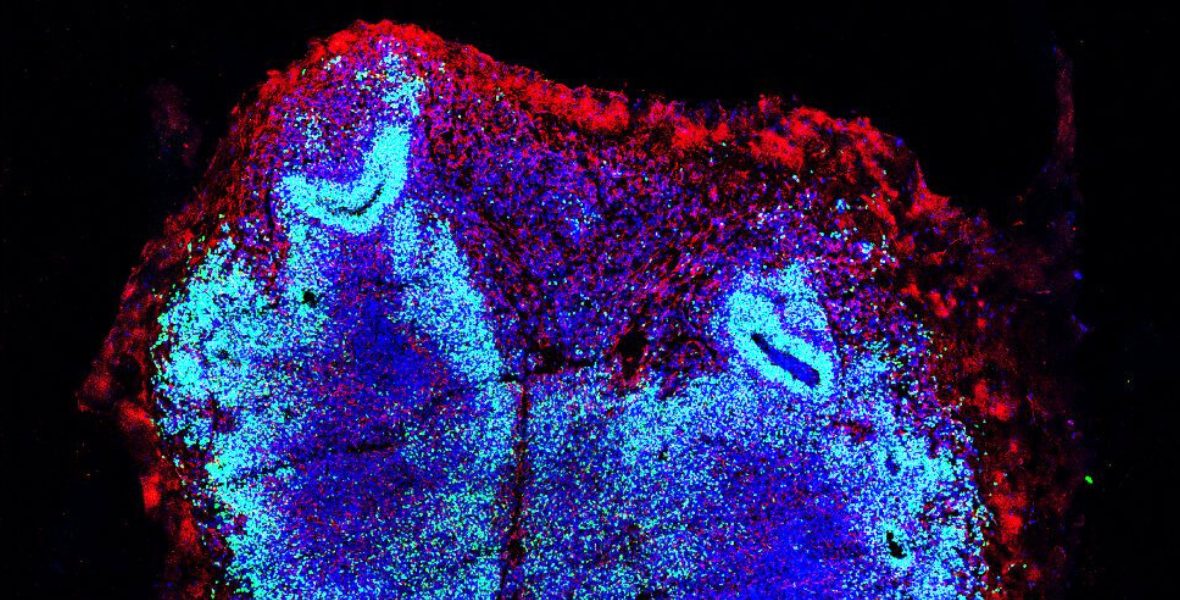
Sezioni di organoidi spinali colorate con marcatori dei progenitori neuronali (in verde), cellule immature del sistema nervoso, e dei neuroni (in rosso), provenienti da individui sani del gruppo di controllo e da pazienti affetti da SMA. I nuclei sono colorati in blu. Le immagini mostrano alterazioni nelle traiettorie di neurosviluppo nella SMA, dimostrando che questa malattia neurodegenerativa è causata da difetti nelle fasi precoci di formazione del midollo spinale e del sistema nervoso centrale in generale. Scala: 200 o 100 μm.
Figura modificata da Faravelli, I., Rinchetti, P., Tambalo, M. et al., Nature Communications 2026.
Il valore dell’intervento terapeutico precoce
Un altro risultato chiave dello studio riguarda l’efficacia del trattamento precoce. Gli autori dimostrano che la somministrazione tempestiva di oligonucleotidi antisenso, terapia progettata per aumentare i livelli di proteina SMN, è in grado di correggere i difetti morfologici e funzionali osservati nelle fasi iniziali dello sviluppo. Il trattamento ripristina pattern di attività elettrica fisiologici e riduce in modo significativo la morte cellulare, fornendo una solida base sperimentale all’importanza dell’intervento prima che il danno neurologico diventi irreversibile.
Tra i senior authors figura anche Monica Nizzardo, ricercatrice del Centro Dino Ferrari, che ha contribuito con la sua esperienza nella ricerca traslazionale sulla SMA. Lo studio è il risultato di un lavoro sinergico che ha coinvolto numerosi ricercatori, tra cui tre giovani scienziate prime autrici: Monica Tambalo (ricercatrice nel gruppo della Professoressa Lodato presso Humanitas University e IRCCS Istituto Clinico Humanitas), Irene Faravelli e Paola Rinchetti, che hanno contribuito in modo determinante allo sviluppo e all’analisi dei modelli di organoidi e all’interpretazione dei dati molecolari e funzionali. La pubblicazione rappresenta il coronamento di una collaborazione pluriennale tra istituzioni di eccellenza nel panorama delle neuroscienze italiane, integrando ricerca di base, clinica e traslazionale.
L’Atrofia Muscolare Spinale resta una delle principali cause genetiche di mortalità infantile, ma le terapie introdotte negli ultimi anni hanno già trasformato la storia naturale della malattia. Questo studio aggiunge un tassello fondamentale alla comprensione dei suoi meccanismi, indicando che i benefici maggiori si ottengono quando il trattamento viene avviato in fase presintomatica, idealmente grazie allo screening neonatale, prima che le alterazioni dello sviluppo del sistema nervoso diventino irreversibili.






